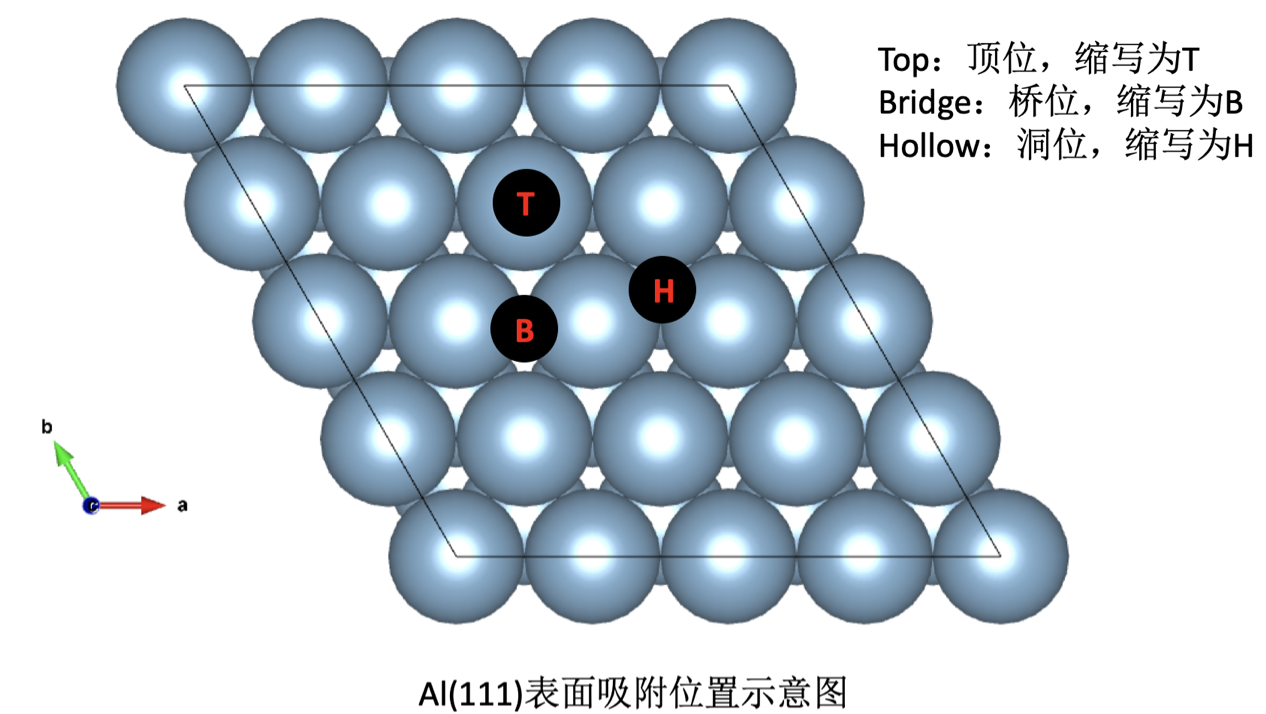
Al4 #代表了体系的名称,第一行随便写。 I'm from China 1.0#缩放系数 4.0389300.0000000.000000 0.0000004.0389300.000000 0.0000000.0000004.038930 Al #体系中元素的名称 4#体系中元素的个数 direct #可以写成分数坐标和直角(Cartersian)坐标,这里面写成分数坐标。 0.0000000.0000000.000000 Al 0.0000000.5000000.500000 Al 0.5000000.0000000.500000 Al 0.5000000.5000000.000000 Al
from pymatgen.core.surface import Slab, SlabGenerator, generate_all_slabs, Structure, Lattice, ReconstructionGenerator, get_symmetrically_distinct_miller_indices from pymatgen.core.structure import Structure from pymatgen.analysis.adsorption import * from pymatgen.ext.matproj import MPRester from pymatgen.symmetry.analyzer import SpacegroupAnalyzer from pymatgen.io.vasp.inputs import Poscar from matplotlib import pyplot as plt import os
formula = struct.composition.reduced_formula print(formula) print("输入的结构是:{}".format(formula))
os.mkdir(formula) os.chdir(formula) ls = get_symmetrically_distinct_miller_indices(struct, 1, return_hkil=False) print(ls)
for i in ls: slab = SlabGenerator(struct, miller_index=i, min_slab_size=25,min_vacuum_size=15.0, lll_reduce=True, center_slab=True) for n, slabs in enumerate(slab.get_slabs(bonds=None, ftol=0.1, tol=0.1, max_broken_bonds=0, repair=False, symmetrize=True)): slabs.make_supercell([[2,0,0],[0,2,0],[0,0,1]]) name = str(i).split(',')[0][1]+str(i).split(',')[1][1]+str(i).split(',')[2][1] open(formula+'_'+name +'_' + str(n+1) + '.vasp', 'w').write(str(Poscar(slabs)))
from pymatgen.io.vasp import Locpot import matplotlib.pyplot as plt import numpy as np
locpot = Locpot.from_file("LOCPOT")
z_locpot = locpot.get_average_along_axis(2)
fermi_level = 1.7968
import matplotlib.pyplot as plt import numpy as np import matplotlib.pyplot as plt from pylab import * #控制刻度线 from matplotlib import cm from matplotlib.collections import LineCollection cm = plt.cm.get_cmap('viridis')